Malattie da Accumulo Lisosomiale (LSD, Lysosomial Storage Diseases): che cosa sono, sintomi e le cure
Le Malattie da Accumulo Lisosomiale sono patologie croniche di origine genetica caratterizzate da una grande eterogeneità di manifestazioni cliniche e insorgono prevalentemente nei primissimi anni di vita, per difetto o assenza di uno degli enzimi contenuti nei lisosomi, organuli presenti allinterno della cellula.
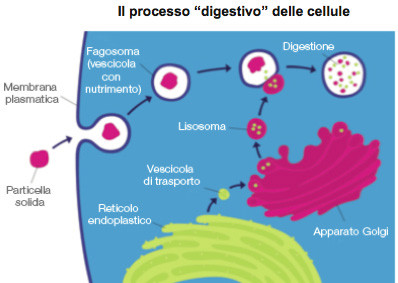
Coinvolti in diversi e complessi processi cellulari come il mantenimento del colesterolo, la riparazione dei tessuti e delle ossa, la difesa da agenti patogeni, la morte cellulare, i lisosomi sono considerati come centri di riciclo della cellula e una delle loro principali funzioni è quella digestiva: la membrana lisosomiale racchiude infatti 50-60 enzimi in grado di digerire e metabolizzare sia molecole di origine intracellulare, sia la gran parte delle macromolecole che sintroducono nelle cellule1,2.
Il difetto o lassenza di uno specifico enzima impedisce il processo di digestione dei prodotti di rifiuto del metabolismo cellulare; a causa di ciò, le macromolecole si accumulano e si espandono allinterno del lisosoma (la cosiddetta costipazione cellulare), determinandone laumento del volume e dunque il danno cellulare e tissutale.
Esistono più di 40 differenti malattie lisosomiali3, ognuna delle quali è associata al difetto o alla carenza di uno specifico enzima. Fra quelle più note:
Malattia di Gaucher (glucocerebrosidasi, detto anche
beta-glucosidasi acida)
Malattia di Fabry (alfa-galactosidasi A)
Sindrome di Hunter o Mucopolisaccaridosi II (iduronato sulfatasi)
Sindrome di Sanfilippo A o Mucopolisaccaridosi III (eparan sulfamidasi)
Malattia di Niemann-Pick (sfingomielinasi)
Malattia di Pompe (alfa-glucosidasi acida)
Malattia di Tay-Sachs (beta-esosaminidasi)
Lorigine di tale difetto o assenza è una mutazione genetica4,5 che porta ad unanomala trascrizione delle informazioni del DNA ai siti della sintesi proteica e dunque alla produzione di enzimi anomali. Il deficit enzimatico allorigine di questa classe di patologie può manifestarsi anche a causa della mancanza di una specifica sostanza in grado di attivare lenzima o per il trasporto difettoso di questo allinterno della cellula, che ne impedisce la corretta localizzazione nel lisosoma1.
In queste malattie, la trasmissione ereditaria avviene attraverso entrambi i genitori portatori sani dellalterazione genetica, con una modalità definita autosomica recessiva: la patologia insorge quando sono alterati entrambi i geni, sui cromosomi materno e paterno. Nella Malattia di Fabry e nella Sindrome di Hunter, invece, lalterazione si trasmette attraverso il cromosoma X.
Queste patologie hanno un decorso progressivo che porta al deterioramento delle funzioni vitali, con esito spesso infausto. Le conseguenze patologiche di tale deficit possono interessare più organi e le manifestazioni cliniche includono lingrossamento del fegato e della milza, la perdita progressiva di funzioni neurologiche, alterazioni degli occhi, del cuore, delle ossa e della muscolatura.
Considerando ciascuna specifica patologia, tali malattie possono essere definite rare e presentano un'incidenza variabile: da 1 caso su 40.000 nascite della Malattia di Gaucher del Tipo 1, a 1 su 100.000 della Malattia di Fabry, per arrivare a 1 su oltre 4 milioni della Sialidosi, il disturbo più raro3,5,6.
Se invece consideriamo le Malattie da Accumulo Lisosomiale nel loro insieme, esse sono relativamente comuni e rappresentano un grave rischio sanitario, avendo unincidenza complessiva di 1 caso su 7.700 nascite3,5; unincidenza che aumenta notevolmente, nello specifico caso della Malattia di Gaucher, tra le popolazioni ebree di origine est-europea, tra le quali tale patologia è particolarmente diffusa e colpisce 1 bambino su 600 - 1.000.
La natura genetica delle Malattie da Accumulo Lisosomiale non consente a tuttoggi la possibilità di una cura definitiva, ma è stato sperimentato con successo lapproccio basato sulla sostituzione dellenzima lisosomiale difettoso, la cosiddetta terapia enzimatica sostitutiva (ERT, Enzyme Replacement Therapy). Questo tipo di trattamento si è dimostrato in grado di alleviare segni e sintomi in alcune delle patologie lisosomiali più diffuse, contribuendo a preservare la qualità di vita dei giovani pazienti che ne sono afflitti.
Note
1. Evans J., Manson A. Crash Course: Cell biology and genetics. Mosby Elsevier, terza edizione, 2008.
2. Saftig P. Physiology of the lysosome, in Mehta A., Beck M., Sunder-Plassmann G. (eds.) Gaucher disease: perspectives from 5 years of FOS, cap. 3, pp. 21-31. Oxford: Oxford PharmaGenesis Ltd, 2006.
3. Meikle P.J., Hopwood J.J., Clague A.E., Carey W.F. Prevalence of lysosomal storage disorders. Journal of the American Medical Association 1999, 281,(3): 249-254.
4. Hopkin R.J., Grabowski G.A. Lysosomal Storage Diseases, in Fauci A.S., Braunwald E., Kasper D.L. et al., (eds.), Harrisons Principles of Internal Medicine, cap. 35, pp. 2452-2456, 17a edizione. The McGraw-Hill Companies Inc, 2008.
5. Mehta A., Beck M., Linhart A., Sunder-Plassmann G., Widmer U. History of lysosomal storage diseases: an overview, in Mehta A., Beck M., Sunder-Plassmann G. (eds), Gaucher disease: perspectives from 5 years of FOS, cap. 1, pp. 1-7. Oxford: Oxford PharmaGenesis Ltd, 2006.
6. MacDermott K.D., Holmes A. and Miners, A.H. Anderson-Fabry disease: clinical manifestations and impact of disease in a cohort of 98 hemizygous males. Journal of Medical Genetics, 2001, 38(11): 750-60.
Fonte: Pro Format Comunicazione Ufficio stampa
