Sindrome di Hunter: che cos'è, sintomi e le cure
La Sindrome di Hunter fa parte della famiglia delle Malattie da Accumulo Lisosomiale, patologie croniche di origine genetica caratterizzate da una grande eterogeneità di manifestazioni cliniche, che insorgono prevalentemente nei primissimi anni di vita, per difetto o assenza di uno degli enzimi contenuti nei lisosomi, organuli presenti allinterno della cellula.
Chiamata anche Mucopolisaccaridosi di tipo II (MPS II), la Sindrome di Hunter prende il nome dal medico canadese Charles Hunter che per primo lha descritta nel 1917. È una malattia rara, progressiva, che colpisce prevalentemente i maschi, con una prognosi a lungo termine estremamente sfavorevole. Sono circa 2.000 gli individui al mondo colpiti dalla Sindrome di Hunter, con unincidenza intorno a 1 caso su 162.000 nati vivi.
La Sindrome di Hunter è caratterizzata dalla deficienza dellenzima iduronato-2-sulfatasi (I2S), che compromette la capacità dellorganismo di degradare e riutilizzare i carboidrati complessi detti glicosaminoglicani (GAG).
Tale deficienza ha come risultato laccumulo di due GAG, il dermatan solfato e leparan solfato, nelle cellule di un gran numero di sistemi e organi: pelle, ossa, articolazioni, cuore, fegato, polmoni, milza e sistema nervoso centrale.
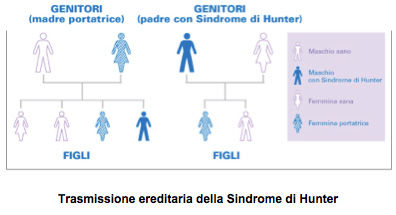
EREDITARIETÀ
La Sindrome di Hunter compare a causa di un difetto del gene I2S sul cromosoma X: a differenza delle donne, gli uomini hanno un solo cromosoma X, per cui I pazienti maschi affetti dalla Sindrome di Hunter generalmente ereditano il gene difettoso dalla madre. Le donne possono ereditare un cromosoma X difettoso anche dal padre, se questi soffre della Sindrome.
Una figlia femmina della stessa madre può essere portatrice, cioè avere un solo cromosoma X con il gene difettoso e trasmetterlo ai figli, oppure può avere due normali cromosomi X. In alcuni casi la Sindrome di Hunter può avere origine senza che sia ereditata dai genitori, a causa di una mutazione del gene I2S durante il processo di formazione del singolo ovocita: in questi casi la madre non è portatrice e il rischio di una mutazione spontanea nei figli successivi è molto basso.
SINTOMI
I primi sintomi della Sindrome di Hunter si manifestano a partire dal secondo anno di età e includono una serie di condizioni patologiche molto frequenti nei bambini, come ad esempio uno stato influenzale persistente, per cui spesso non sono interpretati in modo corretto, procrastinando la diagnosi.
Tra i 18 e i 36 mesi, il bambino manifesta un declino nello sviluppo, con la perdita delle abilità acquisite, spesso accompagnata da sordità. Successivamente, il progressivo accumulo di GAG danneggia i tessuti e sviluppa sintomi più gravi e invalidanti.
Nelle forme gravi, nelle quali la sopravvivenza può non superare i 20 anni di vita, il bambino presenta un addome globoso per epatosplenomegalia disconosciuta e unalterazione dei tratti somatici del volto, e nel primo anno di vita soffre di frequentissime infezioni respiratorie con abbondante produzione di muco che non si riescono a risolvere, ha frequentemente delle ernie inguinali e ombelicali.
ORGANI/APPARATI - SEGNI E SINTOMI
Pelle e capelli
Segni e sintomi: Cute spessa con lesioni nodulari biancastre; capelli ispidi
Testa e collo Tratti facciali grossolani; bocca, lingua e testa allargata; sordità; otiti croniche; frequenti infezioni delle alte vie respiratorie; scarsa visione periferica; cecità notturna
Pelle e capelli
Segni e sintomi: Cute spessa con lesioni nodulari biancastre; capelli ispidi
Testa e collo
Segni e sintomi: Tratti facciali grossolani; bocca, lingua e testa allargata; sordità; otiti croniche; frequenti infezioni delle alte vie respiratorie; scarsa visione periferica; cecità notturna
Torace
Segni e sintomi: Problemi respiratori e ostruttivi; apnea nel sonno; tonsille e adenoidi ingrossate; restringimento tracheale; anormalità della valvola cardiaca e soffio cardiaco; debolezza del muscolo cardiaco; insufficienza cardiaca; aritmie; attacchi cardiaci
Addome
Segni e sintomi:
Ingrossamento del fegato e della milza; ernia; diarrea cronica
Sistema muscolo-scheletrico
Segni e sintomi: Bassa statura; anormalità scheletriche; mani ad artiglio; scoliosi; sindrome del tunnel carpale; rigidezza delle articolazioni; limitato range di movimento
Sistema nervoso centrale
Segni e sintomi: Ritardo mentale e dello sviluppo; perdita delle abilità acquisite; problemi di comportamento; attacchi epilettici; idrocefalia; deficit di attenzione
DIAGNOSI
Interpretare correttamente i numerosi e diversificati sintomi della Sindrome di Hunter non è facile, poiché essi, nella fase iniziale, si sovrappongono a quelli di più comuni condizioni patologiche. È possibile raggiungere una diagnosi certa attraverso la misurazione dellattività dellenzima I2S nel siero, nei globuli bianchi o tramite biopsia cutanea.
Tale analisi è preceduta da esami di laboratorio che possono offrire ulteriori evidenze della presenza della Sindrome; il più utilizzato per lo screening delle MPS è il test delle urine, che verifica un valore elevato di GAG, anche se alcune volte i valori possono risultare normali anche in presenza di malattia. La diagnosi prenatale si esegue misurando lattività enzimatica di I2S nel liquido amniotico o con villocentesi.
TRATTAMENTO DELLA SINDROME DI HUNTER
Non esiste ancora una cura definitiva per questa condizione patologica e prima dello sviluppo nel 2006 della terapia enzimatica sostitutiva (ERT) a base di idursulfasi i pazienti non avevano a disposizione nessun farmaco per contrastare il progredire della sindrome. Lidursulfasi è una copia purificata dellenzima umano iduronato-2-sulfatasi, prodotta con tecnologia del DNA ricombinante: lenzima viene prodotto da una cellula umana nella quale è stato immesso un gene (DNA) che la rende capace di produrre lenzima, che sostituisce quello mancante o insufficiente, riducendo laccumulo dei GAG nelle cellule e rallentando, se non prevenendo, il progressivo danno di tessuti e organi. Idursulfasi è infatti indicato per la terapia a lungo termine ed è in grado di migliorare e controllare efficacemente i sintomi e gli effetti delle complicanze provocate dalla patologia nellorganismo.
Il farmaco è stato oggetto di uno studio della durata di un anno svolto su 96 pazienti di sesso maschile di età compresa tra 5 e 31 anni e lefficacia del medicinale è stata confrontata con quella di un placebo (trattamento fittizio). La sua efficacia è stata provata sulla base della funzionalità polmonare e sulla distanza che i pazienti riuscivano a percorrere camminando in 6 minuti, che consente di misurare gli effetti congiunti della malattia sul cuore, sui polmoni, sulle articolazioni e sugli altri organi.
Idursulfasi ha mostrato di migliorare le funzioni polmonari, con effetti positivi sulla respirazione e la deambulazione dei pazienti che, dopo un anno di trattamento erano in grado di percorrere mediamente 43 metri in più rispetto agli 8 metri dei pazienti trattati con placebo.
Gli effetti indesiderati più comuni, riscontrati in più di 1 paziente su 10, sono le reazioni tipiche correlate allinfusione: reazioni cutanee, febbre, mal di testa, ipertensione e gonfiore nella sede di infusione. Altri effetti indesiderati possono essere affanno, dispnea, dolori addominali, nausea, diarrea e dolori al petto.
La ERT per la Sindrome di Hunter è somministrata ogni settimana sotto forma di infusione, della durata media di tre ore. Recentemente i pazienti che dopo alcuni mesi in clinica tollerano bene le infusioni hanno la facoltà di praticarle a domicilio, grazie al servizio di home care hunter@home, offerto gratuitamente da SHIRE.
Riferimenti: J. Munzer, Overview of the mucopolysaccharidosis, Reumatology 2011; 50:V4-V12
Fonte: Pro Format Comunicazione Ufficio stampa
